![]() 6.5.1 Vibrace molekul
6.5.1 Vibrace molekul
Harmonické přiblížení
V případě obecné víceatomové
molekuly mohou jednotlivé atomy (jejich jádra) vykonávat vůči těžišti molekuly
kmitavý pohyb – vibrace okolo rovnovážných poloh ![]() . Obecně se tedy jedná o soustavu vázaných anharmonických
oscilátorů. V případě malých kmitů, tj. malých odchylek od
rovnovážných poloh, můžeme nahradit potenciál
. Obecně se tedy jedná o soustavu vázaných anharmonických
oscilátorů. V případě malých kmitů, tj. malých odchylek od
rovnovážných poloh, můžeme nahradit potenciál ![]() jeho Taylorovým
rozvojem do členů druhého řádu (harmonické přiblížení):
jeho Taylorovým
rozvojem do členů druhého řádu (harmonické přiblížení):
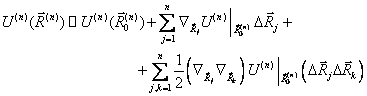
kde ![]() .
.
Rozklad na nezávislé harmonické oscilátory
Vhodnou transformací lze převést potenciál molekuly v harmonickém přiblížení, tj. třetí člen Taylorova rozvoje potenciálu, na
kanonický tvar (diagonalizovat), což znamená, že vypadnou smíšené členy (![]() ):
):
![]()
V hamiltoniánu pro jádra pak musíme provést stejnou transformaci i
v případě operátoru kinetické energie. Ten lze po transformaci formálně
opět zapsat jako výraz![]() , kde parametry
, kde parametry ![]() nejsou rovny původním
hmotnostem atomů a označují se jako efektivní
hmotnosti (srovnej redukovanou
hmotnost v případě dvou částic) myšlených částic – kvazičástic (označují se též jako jednočásticové excitace).
nejsou rovny původním
hmotnostem atomů a označují se jako efektivní
hmotnosti (srovnej redukovanou
hmotnost v případě dvou částic) myšlených částic – kvazičástic (označují se též jako jednočásticové excitace).
V takovém případě je možno v pohybové rovnici pro jádra provést
separaci proměnných: nejdříve separaci pro transformované vektory ![]() a dále separaci pro
tři souřadnice těchto vektorů
a dále separaci pro
tři souřadnice těchto vektorů![]() .
.
![]() , kde
, kde 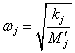 .
.
Transformace souřadnic je obvykle
taková, že jedna z pohybových rovnic popisuje rovnoměrný přímočarý pohyb
těžiště, v tom případě sčítáme v první sumě pouze do n-3. Kvantová čísla ![]() se označují jako vibrační
čísla.
se označují jako vibrační
čísla.
Vibrace dvouatomové molekuly
V případě homonukleární
dvouatomové molekuly (např.
![]() ), ve které konají atomy vibrace ve směru
osy x rovnoběžné se spojnicí jader
atomů, platí pro redukovanou hmotnost
), ve které konají atomy vibrace ve směru
osy x rovnoběžné se spojnicí jader
atomů, platí pro redukovanou hmotnost
![]() a
a
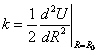 . Zde M představuje hmotnost atomu
a R0 je délka vazby odpovídající
rovnovážné poloze – „nekmitající atomy“.
. Zde M představuje hmotnost atomu
a R0 je délka vazby odpovídající
rovnovážné poloze – „nekmitající atomy“.
Pro frekvenci kmitů tedy máme ![]() . Pro vibrační energii dvouatomové molekuly tak dostaneme vztah
. Pro vibrační energii dvouatomové molekuly tak dostaneme vztah
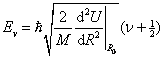 ,
,
kde
![]() je příslušné vibrační
kvantové číslo.
je příslušné vibrační
kvantové číslo.
Vibrace trojatomové lineární symetrické molekuly
| Symetrický vazebný vibrační mód | |
| Asymetrický vazebný vibrační mód | |
| Deformační vibrační mód (dvojnásobně degenerován, obdobná deformace může probíhat v rovině kolmé k nákresně) |
Podle kvantové teorie (viz např. lineární harmonický oscilátor) musí částice i v základním stavu (nejnižším dovoleném energetickém stavu) kmitat s nenulovou kinetickou energií (energie nulových kmitů). U harmonického oscilátoru je střední výchylka částice z rovnovážné polohy nulová pro všechny energetické stavy. U oscilátoru anharmonického, jsou odchylky z rovnovážných poloh malé jen pro nejnižší energetické stavy. Přibližná představa o zaujmutí rovnovážných poloh atomy je tedy přijatelná.